

Avez-vous déjà reçu notre brochure ?

AMS signifie Atrophie Musculaire Spinale. Cette maladie affaiblit les muscles de plus en plus, jusqu’à leur incapacité totale à fonctionner. Plus la maladie survient vite, plus les muscles s’atrophient vite. Fréquemment, les symptômes apparaissent chez les jeunes enfants. Annuellement, environ 10 enfants sur 100000 sont diagnostiqués. Nous pouvons, cependant, leur donner de bonnes nouvelles ! Un des médicaments a été approuvé par l’Agence européenne des médicaments. Et actuellement, un 2ème médicament est en cours d’étude.
La maladie d’AMS est une affection neuromusculaire : il y a un défaut dans les cellules nerveuses qui envoient un signal depuis le cerveau aux muscles. Les cellules nerveuses sont appelées ‘neurones moteurs’. Si les muscles ne reçoivent pas le signal des neurones moteurs, ils ne réagissent pas et ne bougent donc pas. Les muscles qui ne font (presque) plus rien, s’affaiblissent et deviennent tellement minces qu’ils finissent pas ne plus fonctionner du tout. Les muscles commencent alors à s’atrophier. L’amyotrophie attaque surtout les muscles des bras et des jambes. Mais si la maladie se développe trop, les muscles respiratoires aussi peuvent être atteints. Ceci mène à une trop faible oxygénation du corps. A cause de cela, il est possible que l’insuffisance respiratoire chronique requière l’aide d’un appareil améliorant la respiration.
La cause de la maladie de l’AMS provient d’un défaut dans l’un des environ 20000 gènes dont sont dotés les humains. Ces derniers sont de petits morceaux d’ADN qui déterminent l’apparence d’une personne, son caractère ou si elle est en bonne santé. Toute personne possède 46 (23 paires) chromosomes, sur lesquels reposent les gènes. Les scientifiques ont découvert que la cause de la maladie d’AMS se situe sur le 5ème chromosome. Chez les personnes souffrant d’AMS, le gène SMN1 est défectueux ou est totalement absent. Ce gène crée habituellement la protéine SMN, quiconstitue le combustible des neurones moteurs. Sans protéine, les neurones moteurs ne peuvent pas (bien) fonctionner : les signaux du cerveau ne sont pas bien (ou mal) transmis aux muscles. Chez les personnes souffrant d’amyotrophie, le gène SMN2 (une sorte de ‘copie de sauvegarde du gène’ se substitue à la tâche pour créer la protéine SMN. Cependant, le gène SMN ne fabrique que 10 % de protéine utilisable par rapport à ce que fabrique le gène SMN1.
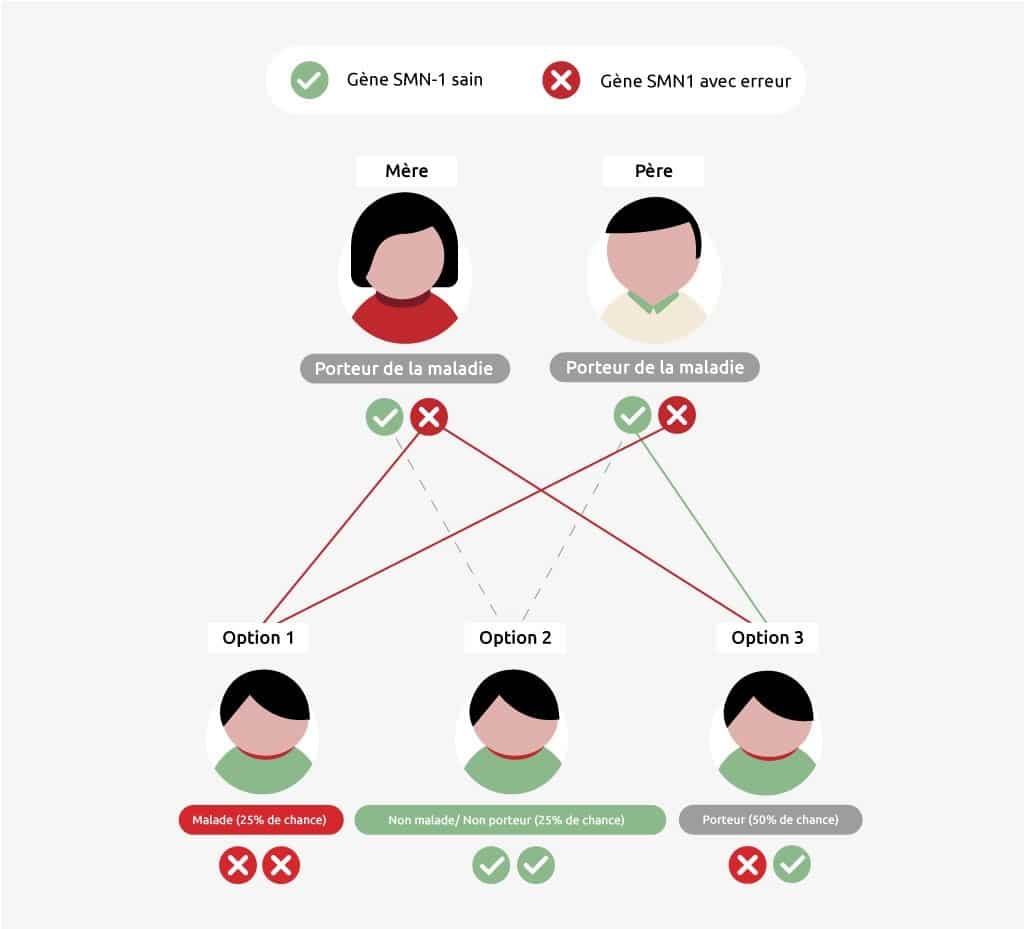
L’AMS est une maladie héréditaire : les enfants reçoivent les gènes endommagés de leurs parents. Si un parent est porteur de la maladie, il dispose d’un gène SMN1 sain et d’un gène endommagé. Le porteur n’est pas malade lui-même, mais il peut transmettre la maladie. Les enfants sont touchés par la maladie uniquement si leur père et leur mère ont transmis un gène SMN1 endommagé ou un 5ème chromosome sans gène SMN1. Le risque qu’un enfant de deux porteurs tombe malade est de 25 %. Le risque que l’enfant ne soit que porteur augmente : il est de 50 %. Ils ont également 25 % de chances de ne pas hériter de la portance de la maladie.
Il existe 4 types d’atrophie musculaire spinale. La distinction entre types se fait en observant l’âge auquel se présente la maladie, et la vitesse d’aggravation des symptômes. Comme la maladie est souvent établie dès la petite enfance, l’amyotrophie peut être considérée comme une maladie infantile. Plus la maladie survient jeune, plus l’évolution est grave.
La différence dans l’évolution de la maladie existe parce que certaines personnes ont plus de ‘copies de sauvegarde’ du gène SMN2 que d’autres. Plus la personne a de ces gènes, plus il peut fabriquer de protéine SMN, et moins la personne sera en manque de cette protéine SMN. Les neurones moteurs pourront donc continuer à fonctionner pendant plus longtemps. Une personne avec 0 à 2 copies développe le type 0 ou 1. Avec 3 copies, cette personne développera le type 1, 2 ou même 3. Avec 4 copies, ou plus, elle développera le type 2 (parfois), 3 ou 4.
Le type 1 de la maladie musculaire d’AMS se déclare chez les bébés, peu après la naissance ou lors des 6 premiers mois de vie. Les muscles de leurs bras, de leur jambes et de leur torse sont souvent affaiblis. Les muscles de l’appareil respiratoire aussi s’affaiblissent. Les bébés atteints de la maladie d’AMS n’arrivent généralement pas à s’asseoir, à lever leur tête ou à se retourner. Ce type est la forme la plus grave de la maladie. Là aussi, l’on peut dire que plus la maladie est développée tôt, plus les symptômes seront graves. Dans certains cas, la maladie peut déjà apparaître avant la naissance, il s’agit alors de l’amyotrophie de type 0. Le type 1 représente environ 55 % des personnes atteintes d’AMS.
Le type 2 de la maladie se déclare auprès des enfants entre 6 et 18 mois. Ils sont surtout gênés par des muscles affaiblis dans les jambes et le dos. Souvent, les bras sont encore assez forts. Les bébés ayant cette forme d’amyotrophie peuvent apprendre à s’asseoir et se retourner, mais ils ne peuvent pas marcher seuls. Les enfants et les adolescents de type 2 ont donc généralement besoin d’un fauteuil roulant. L’espérance de vie pour le type 2 repose entre 10 et 40 ans. Cela peut donc fortement varier d’une personne à une autre.
Si la maladie se présente chez les enfants entre 18 mois et 4 ans, nous parlons du type 3. Les enfants apprennent à s’asseoir et à se lever et à marcher seuls. Il y a pourtant de fortes chances que ces capacités se perdent plus tard en raison de l’affaiblissement de leurs muscles. Les enfants de type 3 aussi devront donc souvent utiliser un fauteuil roulant. Plus tard, dans leur vie, il est même possible qu’ils aient besoin d’un appareil d’aide à la respiration. Avec le type 3 aussi, l’espérance de vie est très variable. Mais les personnes diagnostiquées d’amyotrophie de type 3 ont généralement une espérance de vie normale.

Le type 4 de l’amyotrophie n’existe quasiment pas. Les symptômes se présentent seulement après l’âge de 30 ans. La plupart des gens ayant ce type d’AMS n’ont que très peu de symptômes. Des périodes de détérioration sont suivies de périodes sans symptômes. Les personnes déclarées de type 4 ont souvent une espérance de vie normale. Ils peuvent, néanmoins, avoir des difficultés à monter les escaliers ou à lever les bras, à un âge plus avancé.
Dans la majorité des cas, l’atrophie musculaire spinale est établie par un examen de l’ADN. Un tel examen démontre s’il y a des défauts dans l’ADN. Le type d’AMS ne peut pas être déterminé de cette manière. Il est plutôt déterminé lorsque l’on voit la maladie se déclarer et la vitesse à laquelle elle évolue.
Dans 8 % des autres cas, le défaut du gène ne peut pas être détecté. Il faut alors réaliser un autre type d’examen : un électromyogramme (EMG) ou une biopsie musculaire. Avec un électromyogramme, de fines aiguilles sont enfoncées dans les fibres musculaires. Les aiguilles sont reliées à un appareil d’électromyographie qui mesure les réactions des muscles. Le médecin peut alors voir si les muscles réagissent de façon anormale. Avec une biopsie musculaire, le médecin prélève de petits morceaux du muscle pour les étudier sous un microscope. Il étudie la structure du muscle et fait des essais. Il regarde notamment la taille des fibres musculaires. Si les dimensions sont anormales, cela indique la présence d’une amyotrophie.
Plusieurs médicaments ont été développés pour lutter contre l’amyotrophie et diminuer les symptômes. Pour le moment, le médicament Spinraza a été approuvé en Europe. Par le biais d’une piqûre dans le dos, le gène SMN2 est ainsi stimulé plusieurs fois par an pour créer plus de protéine SMN utilisable. Ainsi, les neurones moteurs bénéficient de plus de combustible à envoyer vers les muscles.
Un autre médicament, appelé Zolgensma, fabrique un nouveau gène SMN1 dans les neurones moteurs, grâce à la thérapie génique. L’avantage est que ce médicament n’a besoin d’être utilisé qu’une seule fois. L’inconvénient est que son coût est de 2 millions d’euros par patient, et nous ne connaissons pas les effets à long terme. En Europe, le médicament a été approuvé pour un usage chez les jeunes enfants. Mais des accords concernant les remboursements du médicament doivent encore être passés avant qu’il ne puisse être réellement utilisé.
Plus sur la santé





